Problems that might occur with any DNA bands (unknowns or standards)
Misshapen bands (a protrusion only 15 to 35% the width of the bands, oriented in a single direction for all the bands)
Examples of misshapen bands with protrusions



Although all precautions have been taken to prevent the appearance of uninterpretable results in this lab, such results are possible if instructions in this protocol are not followed carefully. Generally, because the DNA standards were already digested and mixed with loading buffer, there are fewer problems with the standard bands than with the unknown digests. Also, since the three digest reactions are set up separately, they can vary considerably in overall quality even when set up by the same student. Generally, all three unknown samples should have the same total amount of DNA (300ng if set up correctly) in each lane, whether digested with EcoRI or with BamHI. Although 400 ng of DNA standards should have been loaded in each lane, the standard DNA is divided across 24 kbp of bands or 17 ng/kbp, versus 23 to 40 mg/kbp for the unknowns. This means that the unknown bands should be brighter than any standard band of about the same size.
Some of the most commonly occurring significant problems are described her briefly here. Some of the less significant imperfections are shown in the description of a "perfect gel." Some other oddities that may be observed but are not significant problems include seeing the blue UV bulbs horizontally across the gel picture, the tape or some fluorescent green ink from the marking pen partially obscuring part of your gel, and seeing two dark fuzzy regions running horizontally across the gel (these are shadows caused by the tracking dyes).
Examples of misshapen bands with protrusions
This usually is caused by poking the pipet tip into the side of the well when loading the sample. Ignore the protrusion when measuring the band migration distance for determination of the DNA fragment sizes.
Examples of split bands


This phenomenon is the result of slight variation of the migration rate of the DNA in the well as a function of the distance from the bottom of the gel mold (or top of the gel), causing two "edges" of DNA brightness, one slightly ahead of the other. For some reason, this difference is blurred at the horizontal edges of the band and is only noticable in the middle (on the horizontal axis). This is not usually seen in bands greater than 2KB, and typically is only seen when running the gel fast (170 volts) for substantial portions of the electrophoresis. The biophysical basis for this phenomenon is not understood, but may have to do with slight differences between composition of the gel and the electrode buffer in causing slight variation in the electric field in the gel as a variation of the distance above the gel mold.
If you observe this, remember that these are single bands, even though they appear to be a closely spaced (unresolved) doublet of bands. Also, measure the migration distance to half way between the brightest regions of each edge of the band.
Examples of misshapen bands (wavy, broken, or dumbbell shaped)




When the gel is run fast, the DNA moves more at the sides of the band than in the middle, giving it a shape similar to a dumbbell. This probably is due to pushing the DNA through the gel faster than it can migrate through the gel in a uniform manner, given the drag on its migration by the gel matrix. This effect is greater with loading of larger amounts of DNA, larger sized DNA fragments (which contain more DNA than smaller sized fragments cut from the same plasmid), faster runs, and narrower lanes. As more DNA migrates at the sides of the band, there is more tailing or streaking of the DNA backwards (upwards) towards the wells from the sides of the bands. In severe cases, very little DNA remains in the center of the band giving the band an appearance like a "W" or even appearing to have been split in the middle into two half-width bands migrating in the gel at the same rate. However, the band thickness (viewed from above) usually is not much greater for W-shaped or dumbbell-shaped bands than for rectangular-shaped bands. Though this is a common problem for gels inthis lab, it is not a major problem for data analysis or interpretation. However, when this occurs, the migration distance of such bands should always be measured using the bottom-most point of the band (the part of the band that migrates the fastest). If the band is extremely elongated when viewed from above and is in the shape of a W or is extremely wavy, see the next paragraph about severe tailing and very poor separation.
Examples of severe tailing/streaking from the ends and/or very poor separation


Generally, this appears as severely misshapen bands or bands (including split bands) that are split symmetrically in the middle (shaped like a W). In some casses this band appearanace is associated with slower than normal migration of all the DNA in the gel.
This can be due to have several possible causes, all related to running the gel really fast along with a problem with the composition of the gel and/or samples. If the effect is seen with all the standards and unknowns in a gel, it probably is because of a problem during the casting of the gel. If it only affects only some of the unknowns, it could be due to a problem with the composition of the samples or to something not right in specific subregions of the gel. These are not usually due to loading problems.
If all the bands in the gel show slow migration, extreme tailing, and/or W shapes, this is due to a problem with the general composition of the gel. This is most likely a problem with the agarose concentration or TAE concentration being too high (failure to restore volume or use of TAE instead of water to restore the agarose solution volume).
If only some of the bands have a very irregular shape and/or tail (streaks upward to the well), this can be caused by incomplete dissolution of the agarose before casting the gel (the undissolved agarose blocks DNA migration). Similar effects can result from contamination of the gel with large inert objects (rocks) or crystals of various salts/compounds (such as might be near a balance). Additionally, this can result from problems with the composition of individual unknonw samples, such as being too concentrated (too small a reaction volume due to a mistake) or incorrectly assembled, such as by using BPB or TAE or water instead of 1.25X Reaction buffer and/or enzyme, or use of all enzyme and no reaction buffer.
Extreme cases of this can make analysis of the DNA fragments very difficult. When measuring band migration distances only use the distance from the well to the fastest migrating part of each band rather than the streaked parts of the bands (except for very small sized DNA bands which do not have bright leading edge, in which case use the brighted part in the horizontal center of the band.)
Examples of all the lanes in the entire gel running unevenly from one side to the other
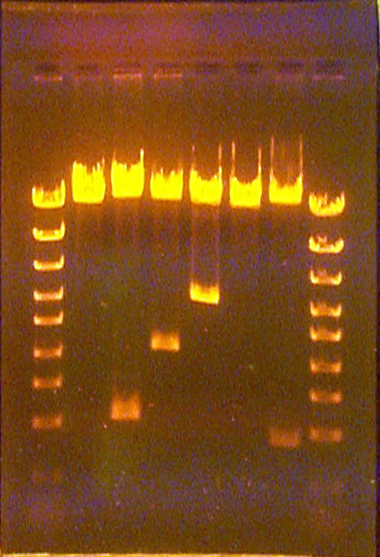
Minor unevenness is not uncommon, but a severe case, as depicted above, is very uncommon. Minor differences in migration across the lanes of a gel can be caused by the gel (in the mold) being slightly at an angle relative to the electrcic field, which is parallel to the long dimesion of the gel apparatus. Usually they make little difference in the size estimation of the unknown bands as long as the migration distances from the closest lane of standards are used to calibrate the function relating size to migration distance. However, in severe cases, which may be caused by the mold being at the largest possible angle to the field in the apparatus, special effort must be exerted to estimate the band sizes correctly.
Since the migration distances of the standards and unknowns are very different when comparing across the entire gel, but appear to change progressively and consistently from lane to lane, it is possible to compensate for this problem and still get fairly accurate estimates for the unknown band sizes. To do this, you must first draw lines across the gel photo that connect the migration measurement points for the two standards of each size (one on each side of the gel). Then, in the verical line that represents the measuring point for each of the unknown lanes, measure the probable migration distances of each standard off these slanted/horizontal lines where they intersect the vertical measuring line (see the figure below). This way, you get a set of standard migration distances for each of the unknown lanes. Use only the probable standard migration distances measured for any one unknown lane for doing the size estimation for unknown bands in that lane. In other words, if you have unknown bands in lanes 5, 6, and 7, create a set of probable standard migration distances for each of lanes 5, 6, and 7, and then, for unknown bands in lane 5, use the standard distances from lane 5 for the size determination. For the unknowns in lane 6, use the lane 6 standard migration distances, etc. Note that in the following example, the topmost unknown band in each gel must have its size determined using extrapolation (see special extrapolation page).
Gel showing how to measure the assumed standard migrations distances for each lane

Examples of RNA cloud at the bottom (below the 193 bp standard)


The DNA samples used in this class have been purified to remove all or nearly all the RNA from the bacteria. Because RNA migrates as a diffuse cloud (due to its non-linear shape) and RNA binds Ethidium more poorly than dsDNA, it tends to be difficult to see even when a large amount is present. Because the new transilluminator provides greater sensitivity of detection than in years past, this year there are a few samples that have enough RNA to be detected. In all cases the RNA appears as a cloud that is much less sharp and much thicker than a DNA band. Also, the RNA cloud appears to migrate faster than the 193 bp DNA standard fragment. Because the RNA is in the unknown DNA samples before the restriction enzyme is added and it is not affected by the digest reaction, it shows up after both the EcoRI and the BamHI digests. Fortunately, only a very few samples have detectable levels of RNA in them.
The RNA contamination should not be considered a DNA band, or else it will interfere with the analysis or interpretation of the data obtained in this lab. RNA, if detected, either may be ignored completely or it may be noted in the lab report and otherwise ignored.
A band of less than 100bp is not likely to be detected using the experimental conditions used in this lab. Furthermore, the smaller the DNA, the lower the chances of detecting it in this lab, since fluorescence of the band is proportional to the amount of DNA in it, and the amount of DNA in a band cut from the same plasmid is proportional to the length of the fragment. In the case of a few samples, RNA contamination of the samples might obscure a very small DNA fragment shorter than 193 bp. This is a limitation of all restriction mapping, and thus mapping of unknown sequences always must allow for the possibility of very small, but undetected, DNA fragments.
Examples of size standards in unknown lanes

This obvious problem has only one cause: Some size standard was added to or leaked into the unknown lane(s) where it is detected. If present in all of the three unknown lanes, then it was probably added instead of 6X loading buffer. When the standard well leaked or overflowed into the adjoining well, it usually only contributes a small amount of DNA to that lane, and so does not obscure the unknown DNA bands.
Addition of standards to an unknown digestion reaction can severely limit the ability to interpret the fragmentation patter of such unknown. Typically, the unknown bands are a bit brighter than the standards, so the unknown DNA bands usually can be identified even if they migrate at the same or nearly the same rate as standard bands. If the unknown sample bands were lighter than normal due to the use of less than 1 microliter of unknown DNA in the digest reaction, then they may be difficult to see relative to the standard bands unless they migrate at a distinct rate. Since each the three reactions was set up separately, the ease of detection of the unknown bands in the presence of contaminating size standards may vary among the three unknown lanes.
Usually, at least some information can be obtained for the unknowns when this problem occurs. Students with this problem are encouraged to do their best to interpret the data from that week, and to rely heavily on the data from the other week of the lab. In cases where the data cannot be reliably and adequately interpreted, the student may request replacement data from Prof. Ashendel. Generally, this will be needed only if the problem occurs during the second week of the lab.
Examples of bands from a neighboring unknown well leaking into an unknown lane
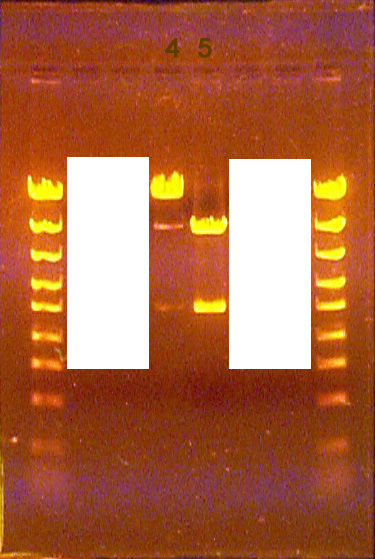
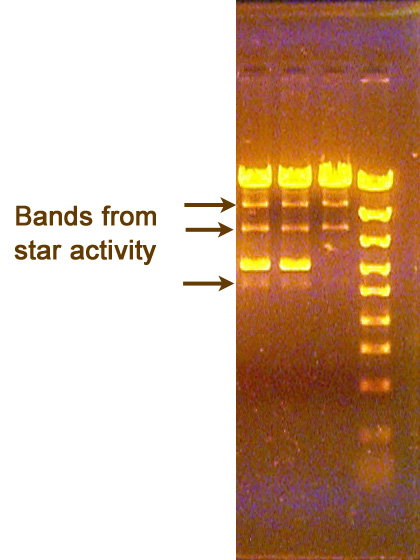
This uncommon problem results from the spilling of one well into the neighboring well. Such contamination is usually at a low level relative to what was loaded into the well, and so the contaminating bands are easily identified by their faint intensity and their migration being identical to the bands in the neighboring lane. Bands that are fainter, but do not migrate the same as either adjoining lane are not likely to be due to contamination from the neighboring well. Rather, such bands may result from incomplete digestion reactions (see below) or from "star activity" of the restriction endonuclease used (also see below).
Examples of no bands in a digest lane
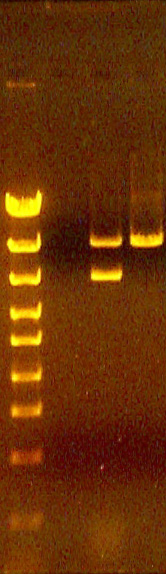
In this lab exercise, this is always caused by failure to add DNA to the digest reaction.
The failure to add DNA to the reaction could have resulted from poor pipetting technique compounded by failure to visually confirm delivery of the DNA solution to the reaction tube. Alternatively, some solution other than the unknown DNA may have been inadvertently delivered to the reaction tube instead of the unknown DNA or the DNA could have been dispensed into the the original unknown tube rather than put into the reaction tube. In all cases this usually does not occur for all three reactions, and students having this problem usually only suffer the loss of data for one unknown sample. However, consistently poor techique can cause a loss of data for multiple samples. Alternatively, it may have resulted from skipping the DNA transfer step or from taking the DNA from the unknown tubes and adding it back into the unknown tubes.
There is a remote possiblity that most of the reaction failed to load into the well or was sucked or washed out of the well, or that it drained out of the well via hole poked in the bottom of the well. Of course, such loss of sample from a well would be obvious because of the lack of tracking dyes in such a lane and such loss should have been noticed and recorded by the experimentor. However, it is unlikely ALL the sampel would be lost, and DNA bands are detectable with just 2% of the total DNA remaining in the lane.
Although not possible in this lab exercise, in a research setting there is the additional possibility that the DNA was completely degraded to nucleotides during the incubatrion by non-specific nucleases that contaminated the reaction (typically produced by microbial growth in a buffer). Given the short reaction time used in this lab and the sterility of the buffers and their storage in a fozen state, the only way the large amount of DNA used in each reaction (60 fmoles, or about 300 ng) could be degraded to a level not able to be detected on the gel (<1 ng) is with an infusion of such a massive amount of bacteria that it is impossible for this to occur in this lab - even when no care was taken to prevent microbial contamination.
Normally, the lack of data from a single sample is not a problem if it happens during the EcoRI mapping done during the first week of lab. This is because the mapping results from the other two samples, along with the information given about the nature of the three unknown DNA samples, makes it possible to guess with a high degree of certainty about the nature of the missing data. During the second week of lab, there is a chance that the lost data will be insignificant (i.e., vector only), or it could be highly significant. If the lost data are essential for analysis/interpretation of the results, Prof. Ashendel will provide the data that you should have obtained.
Examples of only very faint bands in a digest lane
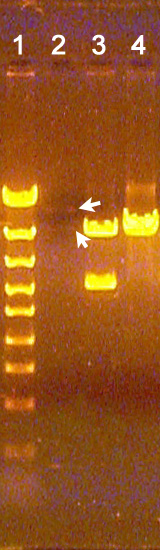
This is similar to the situation described immediately above, where no DNA bands are observed in an unknown lane. However, because one or more very faint bands are observed, it is more possible that this has resulted from wash out of sample from the well or misloading of the sample into the well (though this should have been noticed). This also may be a combination of spillover from the adjacent lane and failure to add any DNA to the reaction tube.
Though identifying the cause of this is more difficult, the good news is that the faint bands detected may be useful for analysis and interpretation of the unknowns. Clearly, if they result from contamination, the results are not useful. Also, if a very small fragment is predicted to be present and its detection is critical for interpretation of the results, this problem probably will make it impossible to detect such a small fragment.
There is more information available about incomplete digestions and star activity
The top-most band in the three EcoRI digests did not migrate to the same position in the gel
Assuming that the DNA bands in the three unknowns are about the same brightness (i.e., there was not significant loss of one or more samples), then the only explanation for this is incomplete digestion in one or more of the three unknown digestions. This cannot be caused by enzyme "star" activity. See above for examples and information about incomplete digestions.